









Very virulent infectious bursal disease virus: reduced pathogenicity in a rare natural segment-B reassorted isolate
1 Cyril Le Nouen, 1 Gaelle Rivallan, 1 Didier Toquin, 2 Pierre Darlu, 3 Yannick Morin, 4 Ve´ ronique Beven, 4 Claire de Boisseson, 5 Christophe Cazaban, 5 Sylvain Comte,5 Yannick Gardin and 1 Nicolas Eterradossi 1,3,4 French Agency for Food Safety (AFSSA), Avian and Rabbit Virology, Immunology and Parasitology Unit, OIE Reference Laboratory for Infectious Bursal Disease1, Experimental Services for Avian Pathology (SEEPA)3 and Virus Genetics and Biosecurity Unit 4, BP 53, 22440 Ploufragan, France 2 INSERM U535 Genetic Epidemiology and Structure of Human Populations, 94817 Villejuif Cedex, France 5 CEVA-sante´ animale, BP 126, 33501 Libourne Cedex, France 30 June 2009
30 June 2009
 32 minute read
32 minute read
The purpose of this study was to compare the molecular epidemiology of infectious bursal disease virus (IBDV) segments A and B of 50 natural or vaccine IBDV strains that were isolated or produced between 1972 and 2002 in 17 countries from four continents, with phenotypes ranging from attenuated to very virulent (vv). These strains were subjected to sequence and phylogenetic analysis based on partial sequences of genome segments A and B. Although there is co-evolution of the two genome segments (70% of strains kept the same genetic relatives in the segment A- and B-defined consensus trees), several strains (26 %) were identified with the incongruence length difference test as exhibiting a significantly different phylogenetic relationship depending on which segment was analysed. This suggested that natural reassortment could have occurred. One of the possible naturally occurring reassortant strains, which exhibited a segment A related to the vvIBDV cluster whereas its segment B was not, was thoroughly sequenced (coding sequence of both segments) and submitted to a standardized experimental characterization of its acute pathogenicity. This strain induced significantly less mortality than typical vvIBDVs; however, the mechanisms for this reduced pathogenicity remain unknown, as no significant difference in the bursal lesions, post-infectious antibody response or virus production in the bursa was observed in challenged chickens.
INTRODUCTION
Details of the strains used in this study and the oligonucleotide primers used are available as supplementary material in JGV Online.
Infectious bursal disease virus (IBDV) is a pathogen of worldwide importance to the poultry industry. IBDV destroys B lymphocytes in the bursa of Fabricius in young chickens, causing both mortality and immunosuppression (Lasher & Shane, 1994). Very virulent (vv) IBDV strains emerged in Europe in the late 1980s, causing up to 60% mortality (Chettle et al., 1989; van den Berg et al., 1991).
IBDV belongs to the family Birnaviridae, genus Avibirnavirus, and has a bisegmented dsRNA genome (Delmas et al., 2004). Genome segment A (3?2 kbp) encodes in a major open reading frame (ORF) a precursor polyprotein which is cleaved by autoproteolysis to yield mature VP2 (outer capsid), VP4 (protease) and VP3 (inner capsid) (Kibenge et al., 1988). Segment A also encodes, from a small ORF which precedes and partially overlaps the large ORF, a non-structural protein, VP5 (Spies et al., 1989), possibly involved in virus release (Lombardo et al., 2000; Yao & Vakharia, 2001). Genome segment B (2?8 kbp) encodes the virus polymerase, VP1 (Macreadie & Azad, 1993), the polymerase activity of which has recently been characterized unequivocally in vitro (von Einem et al., 2004).
Molecular characterization of IBDV has been based mainly on the study of the VP2 gene, the middle third of which contains a variable region (vVP2) (Bayliss et al., 1990) where stretches of hydrophilic amino acids have been recognized as the molecular basis for antigenic variation (Schnitzler et al., 1993; Vakharia et al., 1994). Several amino acids in vVP2 have also been proposed as putative markers for pathogenic IBDV. First, amino acid alignments of vVP2 in classical or vvIBDV isolates revealed that most of the latter shared four conserved amino acids, A222, I256, I294 and S299 (Brown & Skinner, 1996; Eterradossi et al., 1999). These positions have since been used as the target of several molecular or antigenic tests aimed at the presumptive molecular or antigenic identification of putative vvIBDVs (Eterradossi et al., 1998; Zierenberg et al., 2000). Second, using the reverse-genetic system developed for IBDV (Mundt & Vakharia, 1996), it was recently demonstrated that vVP2 mutations Q253H, D279N and A284T are involved in both cell-culture adaptation and attenuation (Lim et al., 1999; Mundt, 1999; Brandt et al., 2001; van Loon et al., 2002). Altogether, the molecular basis for pathogenicity of IBDV is not yet fully understood. Indeed, other reverse-genetics studies showed that mosaic classical virus containing VP2 of a vvIBDV strain induced neither morbidity nor mortality in young chickens (Boot et al., 2000), thus demonstrating that VP2 was not the sole determinant for virulence. In addition, other studies have also suggested a role for VP5 (Yao et al., 1998) or suggested, for a limited number of strains, that the B segments of vvIBDVs might also be genetically related. This finding supports the hypothesis that both segments may be involved in pathogenicity (Islam et al., 2001), as also suggested by data on laboratory-generated chimeric viruses which showed segment B to be involved in the efficiency of virus replication (Zierenberg et al., 2004; Liu & Vakharia, 2004; Boot et al., 2005).
The purpose of this study was to compare the molecular epidemiology of IBDV segments A and B of a wider panel of natural or vaccine IBDV strains that were isolated or produced in several countries over a long period and exhibited phenotypes ranging from attenuated to vvIBDV. These strains were submitted to sequence and phylogenetic analysis based on partial sequences of genome segments A and B. We showed that, although there is generally co-evolution of the two genome segments (70% of IBDV strains kept the same phylogenetic relatives in both the segment A- and segment B-derived trees), several strains (26 %) were identified as exhibiting significantly different genetic relatives depending on which segment was analysed. This suggested that natural reassortment is likely to have occurred. One of the possible naturally occurring reassortant strains (02015.1), which exhibited a segment A related to the vvIBDV cluster whereas its segment B was not, was sequenced more thoroughly and was submitted to a standardized experimental characterization of its acute pathogenicity.
METHODS
Virus strains in the phylogenetic study. Fifty IBDV isolates exhibiting various degrees of virulence (vvIBDV, classical, attenuated) were collected from 17 countries and four continents from 1972 to 2002 (Supplementary Table S1, presenting references for the geographical origin and sources of the isolates, is available in JGV Online). Viruses were propagated in chicken embryo fibroblasts (CEF) for the cell-culture-adapted strains or in specific-pathogenfree (SPF) chickens for the non-adapted strains.
Virus strains in the experimental study. IBDV strains 89163 (Eterradossi et al., 1992) and Faragher 52/70 (Bygrave & Faragher, 1970) were used as reference strains typifying vvIBDV and classical IBDV strains, respectively. Strain 02015.1 (vvIBDV-like segment A, unrelated segment B) was selected for experimental assessment of acute pathogenicity. Virus suspensions, produced as described previously from the bursae of inoculated chickens (Eterradossi et al., 1992), were used as inocula. Inocula were titrated by inoculating serial tenfold dilutions (0?1 ml per egg, via the chorioallantoic membrane using seven eggs per dilution) into 9- to 10-day-old SPF eggs (AFSSA-Ploufragan). IBDV titres were calculated according to the method of Reed & Muench (1938) and expressed as median embryo infectious dose (EID50). Absence of reovirus or adenovirus contaminants in the inoculated field strain was assessed by three serial passages in SPF chicken liver embryonic cells. The absence of other avian pathogens potentially present in the inoculum was assessed by serological testing of the sera collected 27 days post-inoculation (p.i.) of SPF chickens. The following antigens were studied: reovirus strain 1133, fowl adenovirus group 1 serotype 1 (CELO virus), infectious bronchitis virus, infectious laryngotracheitis virus, turkey rhinotracheitis virus, the viruses of Marek’s disease (antigen A), Newcastle disease, egg-drop syndrome and chicken anaemia and influenza virus.
Partial amplification and sequencing for the phylogenetic study. Virus RNA was extracted from the bursa of Fabricius of inoculated chickens or from infected CEF monolayers. The sequences of the oligonucleotides used in this study are presented in Supplementary Table S2. The antisense primers were used for reverse transcription (RT). For the PCR, pairs of conserved primers defining approximately 600 bp PCR products were selected in genome regions flanking either the hypervariable domain of the VP2 gene in segment A (nt 744–1180; sequence of primers as in Eterradossi et al., 1998) or in the 59 two-thirds of segment B (nt 297–1749). These regions have been identified previously as the more phylogenetically representative parts of segments A and B, respectively (Le Noue¨n et al., 2005). To facilitate sequencing of the RT-PCR products, chimeric oligonucleotide primers were defined by coupling the sequence of the M13 and 21M13 standard primers to the 59 end of the IBDV-specific sense and antisense primers, respectively. The resulting primer pairs allowed the determination of 514 or 517 bp in the segments A of serotype 1 or 2 strains, respectively, and of 544 bp in the segment B of all studied strains. RT-PCRs, purification of RTPCR products and sequencing were performed according to previously published protocols (Eterradossi et al., 1998, 1999), except for the use of the HEF proofreading enzyme (Roche) which was used instead of Taq polymerase at the PCR step. The resulting nucleotide sequences were submitted to the EMBL database with the accession numbers shown in Supplementary Table S1.
Phylogenetic analysis of the A and B partial sequences. The nucleotide sequences were aligned using CLUSTAL W and were then analysed using the PHYLIP (version 3.57c; Felsenstein, 1993) and PhyML (version 2.1b1; Guindon & Gascuel, 2003) suites of programs. The aligned sequences were submitted to bootstrap (500 replicates; SEQBOOT). The bootstrap-generated datasets were then analysed by the neighbour-joining (NJ) method (DNADIST with Kimura two-parameter distances, followed by NEIGHBOR), the parsimony method (P) (DNAPARS) and the maximum-likelihood (ML) method (PhyML, using the HKY model with optimized invariant sites and transition to transversion ratios). The Jasper strain of infectious pancreatic necrosis virus (genus Aquabirnavirus) was used as an outgroup in all phylogenetic analysis (GenBank accession numbers M18049 and M58756 for segments A and B, respectively). Consensus trees for both regions were determined with CONSENSE. Only groups defined by bootstrap values greater than 80% with all of the NJ, P and ML methods were considered as significant groups.
Congruence between the A and B phylogenies. This was tested with the incongruence length difference (ILD) test (Farris et al., 1994), using the PAUP* program (Swofford, 2002). The ILD test evaluates the null hypothesis that two datasets A and B describing two different characters of the same strains are congruent, i.e. these datasets, when combined, produce a parsimonious tree with a length statistically comparable to the sum of the lengths of the most parsimonious trees obtained from the separate data. To do so, the program calculates the lengths of the most parsimonious trees as measured from the A and B datasets (LA and LB, respectively), as well as LA+B from the (A+B) dataset combining the A and B characters. The test calculates ILDAB=LA+B2(LA+LB). To test the null hypothesis of congruence, two datasets (X and Y) are drawn at random from the combined dataset (A+B), each set having the same size as the original A and B sets, respectively. The value of ILDXY test is then calculated for a large (e.g. 1000) number of such random partitions. When ILDAB exceeds a proportion of 95 or 99% of the ILDXY values, the null hypothesis of congruence can be rejected at P<0.05 or 0.01, respectively. As an iterative process, serial removal of the same strain from the A and B datasets (and studying how the ILDAB index varies) allows the determination of how critically this strain contributes to the incongruence of the phylogenies derived from A and B (Lecointre et al., 1998).
In the present study, the parsimonious trees were obtained using PAUP* (Swofford, 2002) through heuristic procedures with random sequence addition and tree bisection–reconnection (TBR) branch-swapping algorithms. The distribution of ILDXY was assessed on 1000 resampled datasets. The 0?05 probability threshold was taken as significant incongruence. One or a minimal combination of the studied IBDV strains were iteratively removed from the A and B datasets, until the probability of the ILDAB index was greater than 0?05, hence allowing the null hypothesis of congruence between the A and B datasets to be accepted.
Full-length amplification and sequencing of genome segments A and B of a selected strain. Strain 02015.1 was selected following the phylogenetic study as having significantly different phylogenetic relatives in segments A and B. In order to test the hypotheses of (i) a recombination event occurring within segment A or B of 02015.1 and (ii) other specific genetic changes affecting this strain, we sequenced the full coding sequence of both genome segments of strain 02015.1 from overlapping RT-PCR products (same protocol as above; primers as shown in Supplementary Table S2).
The full-length coding sequences of segments A and B (nt 188–3103 and 131–2752; GenBank accession numbers AJ879932 and AJ880090, respectively) of strain 02015.1 were used to determine their genetic relatives, as described above, by NJ, P and ML phylogenetic analysis, based on a panel of homologous IBDV sequences retrieved from databases that had been used to analyse the same regions in a previous study (Le Noue¨n et al., 2005).
Experimental study of the pathogenicity of the potential reassortant virus. One hundred 6-week-old SPF white leghorn chickens (AFSSA-Ploufragan) were housed in four groups of 25 birds of comparable sex and weight, in separate filtered-air, negativepressure isolation units. Chickens in each group were blood sampled for serological testing on the first day. On the same day, each bird in three of the four groups was inoculated by the intranasal route with 105 EID50 of either strain 89163, strain Faragher 52/70 or strain 02015.1; the remaining group was kept as a mock-inoculated control receiving PBS only. Cumulative mortality rates at 5 days p.i. were measured. At 4, 13 and 27 days p.i., at least five birds per group were weighed, humanely killed and necropsied and their bursae were weighed for calculation of the bursa to body weight (b/B) ratio. Each bursa was then cut in two halves; one half was kept for histological examination and the other was processed for preparation of a virus suspension (see below). All surviving birds at 27 days p.i. were blood sampled for final serological examination.
Histological examination. Histological examination was performed by Dr M. Lagadic (Maisons-Alfort, France). Sixty-one bursae of Fabricius from the four different groups (mock-inoculated group and the three inoculated groups) and from three sampling times (4, 13 and 27 days p.i.) were examined individually. The severity of the IBD-induced lesions was quantified according to Skeeles’ scale (Skeeles et al., 1979), which is based on five degrees: 0, no lesions; 1, mild scattered cell depletion in a few follicles; 2, moderate, one-third to half of the follicles having atrophy or depletion of cells; 3, diffuse, atrophy of all follicles; 4, acute inflammation and acute necrosis typical of IBD.
IBDV neutralization test. Virus neutralization (VN) tests to detect antibodies induced by IBDV were performed in CEF cells, using 100 median tissue culture infective doses (TCID50) per well of the CT (serotype 1) IBDV strain, as described previously (Eterradossi et al., 1997a). The VN titre was expressed as log2 of the last dilution resulting in 100% neutralization of the cytopathogenic effect. VN titres greater than 3?3 log2 were considered as positive.
Virus titres in bursae collected at 4 days p.i. Virus suspensions were prepared separately from each half-bursa collected at 4 days p.i., according to previously published protocols (Eterradossi et al., 1992) and based on a standardized organ-to-buffer dilution ratio (1 g to 2 ml). For each of the three challenge viruses, the five virus suspensions obtained after processing of the five birds studied at 4 days p.i. were pooled in equal amounts and mixed. The three pools (one per inoculated virus) were titrated as described above in 10-day-old embryonated SPF eggs.
Amounts of IBDV antigen in bursae collected at 4 days p.i. Antigen capture- (AC-) ELISA was performed as described previously (Eterradossi et al., 1997a, b). Briefly, 96-well polystyrene ELISA plates were coated with a chicken monospecific polyclonal anti-F52/70 serum diluted in PBS. Blocking was performed overnight in blocking buffer. Serial twofold dilutions of the individual virus suspensions prepared at 4 days p.i. were incubated in the coated plates and captured IBDV particles were detected with a reference mouse anti-F52/70 antiserum, followed by an anti-mouse alkaline phosphatase conjugate. Absorbances measured for the serially diluted individual virus suspensions were plotted as dose–effect curves.
Statistical analysis of results. The x2 test was used to compare percentage mortality measured for 5 days p.i. The Kruskal–Wallis one-way analysis of variance was used to compare body weights measured at day 0, b/B ratios, histological scores, levels of virusneutralizing antibodies and amounts of IBDV antigen detected by AC-ELISA in the bursae of inoculated chickens, as measured in the four groups.
RESULTS
Molecular epidemiology of IBDV segments A and B
The phylogenetic study of segment A revealed nine significant clusters (bootstrap value ¢80% in NJ, P and ML) (Fig. 1a). Cluster (i) grouped Ivorian-like pathogenic IBDVs (four isolates), which proved significantly related to cluster (ii), grouping European-like vvIBDVs (23 isolates). Cluster (iii) contained European classical pathogenic viruses (two isolates), whereas clusters (iv) and (v) grouped most vaccine strains (six strains). Cluster (vi) grouped US antigenic variants (three isolates), cluster (vii) grouped vaccine strains (two strains), cluster (viii) grouped the early European strains (two isolates) and cluster (ix) grouped four Australian isolates. The phylogenetic study of segment B proved highly congruent with that of segment A, as six out of nine segment-A-defined significant clusters, clusters (i), (ii), (iii), (v), (vii) and (viii), also proved significant in the segment-B-defined consensus tree (Fig. 1b). Of 46 IBDV isolates exhibiting significant segment-A-defined genetic relationships (all strains except 228E, 05/5, 23/82 and TY89), 32 isolates (70 %) kept the same genetic relatives in the segment-B-defined consensus tree. Moreover, segments B of four Australian strains [group (ix) defined in segment A phylogeny] proved phylogenetically related with the NJ and P methods (bootstrap value >80 %) but not with the ML method (66% bootstrap value) (data not shown).
The ILD test was used to identify potential naturally occurring reassortant strains. The ILDtest revealed that the two trees were not congruent (P¡0?05) unless strains GX, Henan, 02015.1, 02015.3, 02015.2, TY89, 23/82, 05/5, Cu1WT, 78GSZ, 80GA, Bursine2 and Bursine [13 strains (26 %); four groups] were removed. When all of these strains were removed from both phylogenies, the congruence hypothesis could not be rejected (P=0?37), and removing more strains did not change this result. Among the 13 incongruent strains, five were especially interesting with regard to the hypothesis that possible reassortment involving vvIBDV had occurred. Indeed, strains GX, Henan, 02015.1 and 02015.3 exhibited a segment A significantly related to the vvIBDV cluster (bootstrap value of 100 %), whereas their B segments, although they exhibited some differences, all proved significantly different from the viruses belonging to the segment-B-defined clusters (i) or (ii) (vvIBDV-related strains) or to the viruses belonging to clusters (iii), (v), (vii) and (viii) (classical, vaccine, early European and serotype 2 strains). Similarly, strain 02015.2 exhibited a segment A belonging to the vvIBDV cluster but a segment B more significantly related to classical, vaccine or early European viruses [clusters (iii), (v), (vii) and (viii), respectively].
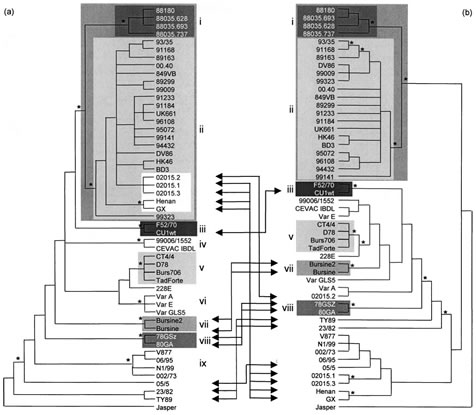
Fig. 1. Phylogenies obtained by the parsimony method of IBDV segments A (a) and B (b). Groups of strains with bootstrap values greater than 80% with the P, NJ and ML methods and present in both segment A and B phylogenies are represented by shading. Arrows indicate movements of strains or group of strains which exhibited different genetic relatives for segments A and B. Asterisks indicate nodes with bootstrap values greater than 80% with the P, NJ and ML methods but possibly not in both phylogenies.
Amplification, sequencing and phylogenetic analysis of the full-length coding regions of segments A and B of the 02015.1 isolate
The coding regions of segments A and B (nt 188–3103 and 131–2752, respectively) of the 02015.1 isolate were sequenced and compared with the corresponding consensus sequence of the typical vvIBDV strains HK46 (GenBank accession numbers AF092943 and AF092944), BD3 (AF36- 2776 and AF362770), D6948 (AF240686 and AF240687) and UK661 (X92760 and X92761). This revealed two segment- A-encoded amino acid changes, one in VP5 (position 135; T in vvIBDV and S in 02015.1, abbreviated as T135S) and one in VP4 (P613S). In genome segment B, mutations encoding 13 amino acid changes were spread along the VP1 gene (I61V, D146E, N147S, T236S, E242D, M274V, M390L, V557I, Q561H, P562S, T576A, R695K and T859I). Phylogenetic analysis of the 02015.1 sequences with their counterparts in a panel of vvIBDV and classical strains retrieved from databases (Le Noue¨n et al., 2005) produced results that were highly consistent with the phylogenies previously obtained for the partial sequences of the studied segments (data not shown). Indeed, the coding region of the A segment of 02015.1 proved significantly related to the vvIBDV cluster (100% bootstrap value with the NJ, P and ML methods), whereas the coding region of the B segment of the same strain branched out of the vvIBDV group in 95, 99 and 89% of the bootstrap-generated trees with the NJ, P and ML methods, respectively.
Pathogenicity of the possible naturally reassortant 02015.1 isolate
On the first day of the experiment, the mean weight of the chickens in the four groups ranged from 440 to 463 g and did not prove to be significantly different (Table 1). Following inoculation, neither disease signs nor mortality were observed in the mock-inoculated control group. In the groups receiving the challenge viruses, morbidity and mortality was observed on days 3 to 5 p.i. The typical vvIBDV strain 89163, the classical strain F52/70 and isolate 02015.1 induced 100, 32 and 24% morbidity and 52, 24 and 8% mortality, respectively. The x2 test showed that mortality induced by the 89163 virus was significantly higher than that observed following challenge with F52/70 or 02015.1, which were not significantly different (Table 1).
The study of b/B ratios at 13 days p.i. revealed comparable bursal atrophy in the three challenged groups (with mean b/B ratios ranging from 0.9 to 1.1 %), which proved statistically significant compared with mock-inoculated controls (mean b/B ratio 3.7 %; P<0.05). The same level of atrophy was still apparent in the three challenged groups at 24 days p.i. (mean b/B ratio in the three inoculated groups 0.6 % versus 3.2 % for the controls). Histological examination of the bursae revealed no lesions in the mockinoculated control group and extensive lesions at 4, 13 and 24 days p.i. in the three inoculated groups. The mean lesion scores of the three latter groups did not differ significantly, irrespective of the examination date (Table 1).
VN titres were measured at 24 days p.i. and revealed high anti-IBDV antibody titres in the three inoculated groups, whereas no neutralizing antibodies were detected in the SPF controls. No significant difference in antibody levels was observed in birds inoculated with 02015.1, 89163 or F52/70 (geometric mean VN titres of 12.3 log2 ± 1.5, 12.5 log2 ± 1.4 and 12.4 log2 ± 1?0, respectively).
On the last day of the experiment, the mean weight of the chickens revealed a statistically lower mean weight in the three challenged groups (mean weights of 979–983 g) compared with the mock inoculated group (mean weight 1116 g; P<0.05). However, the weights of the three inoculated groups did not differ significantly (Table 1).
Virus production in the bursae of infected chickens
Virus titres in the pools of bursal homogenates collected at 4 days p.i. were 105?88, 105?64 and 105?67 EID50 ml21 for viruses 02015.1, 89163 and F52/70, values not considered as different. Similarly, the amounts of IBDV antigen in the bursae at 4 days p.i. were determined after 02015.1, 89163 and F52/70 inoculation. Dose–effect curves obtained in AC-ELISA from serially diluted individual virus suspensions did not exhibit any differences, as tested with the Kruskal–Wallis test (data not shown).
| Table 1. Pathogenicity of IBDV isolate 02015.1 compared with the reference vvIBDV strain 89163 and classical strain F52/ 70 and mock-inoculated controls Data are presented as means ± SD (minimum–maximum) [number of individuals]. Within a row, values indicated by the same superscript letter do not differ significantly according to the x2 test (mortality) or the Kruskal–Wallis test (other parameters) at the P<0.05 confidence level. |
||||
| Parameter | Mock-inoculated | 02015.1 | 89163 | F52/70 |
|---|---|---|---|---|
| Initial weight (g) | 458±61a (376–574) [25] | 440±47a (355–530) [25] | 463±48a (394–559) [25] | 451±45a (389–558) [25] |
| Mortality at 5 days p.i. (%) | 0 | 8a | 52b | 24a |
| b/B ratio (%) 4 days p.i. 13 days p.i. 27 days p.i. |
4.2±0.4a (3.8–4.7) [4] 3.7±0.7a (3–4.8) [5] 3.2±0?9a (1.5–5.1) [15] |
2.9±0.8b (1.9–3.9) [5] 0.9±0.2b (0.7–1.1) [5] 0.6±0.2b (0.1–0.9) [15] |
3.1±0.3b (2.7–3.5) [6] 1±0.2b (0.8–1.2) [5] 0.6±0.1b (0.4–0.7) [7] |
4.4±1.2a (2.6–5.8) [7] 1.1±0.3b (0.7–1.4) [5] 0.6±0.2b (0.3–1) [13] |
| Bursal lesion score* 4 days p.i. 13 days p.i. 27 days p.i. |
0±0a (0–0) [5] 0±0a (0–0) [5] 0±0a (0–0) [5] |
4±0b (4–4) [5] 3±0b (3–3) [5] 2.8±0.5b (2–3) [5] |
4±0b (4–4) [5] 3±0b (3–3) [5] 3±0b (3–3) [4] |
4±0b (4–4) [6] 3±0b (3–3) [4] 3±0b (3–3) [5] |
| VN titre (log2) at 27 days p.i. | 2.3±0a (2.3–2.3) [15] | 12.3±1.5b (10–15) [15] | 12.5±1.4b (11–15) [7] | 12.4±1.0b (11–4) [13] |
| Final weight (g) at 27 days p.i. | 1116±155a (914–1321) [15] | 983±130b (742–1199) [15] | 980±78b (871–1078) [7] | 979±107b (850–1178) [13] |
*Determined by histological grading according to Skeeles et al. (1979) as outlined in Methods.
DISCUSSION
Our phylogenetic studies have shown strong sequence conservation of both segments A and B in different genetic clusters of IBDV and not only in vvIBDVs. These results suggest co-evolution of the two genome segments. This could be due partly to the geographical clustering of some of the studied isolates: Australian IBDV have already been shown to evolve ‘on their own’, probably due to the geographical isolation of the Australian continent (Ignjatovic & Sapats, 2002), as already described for Australian strains of infectious bronchitis virus (Sapats et al., 1996). Similarly, the coevolution could stem from the temporal clustering of some isolates, such as the early European strains 78GSz and 80GA, which have been suggested to represent a European IBDV lineage possibly already established prior to the emergence of the ‘F52/70-like’ classical IBDVs (Domanska et al., 2004). However, co-evolution of genome segments as a major evolutionary feature may still appear surprising in IBDV, as co-infection (and hence reassortment) is likely to be encouraged in the field when some flocks infected by pathogenic IBDVs possibly receive live classical vaccines while incubating the disease. The observed co-evolution therefore suggests that some mechanisms reduce the generation or successful transmission of reassortant viruses: possible mechanisms controlling segment compatibility could include interactions between the segment-A-encoded VP3 and segment-B-encoded VP1 (Tacken et al., 2000, 2002) or between VP1 or VPg and the genome segments (Mu¨ller & Nitschke, 1987). Our results also confirmed previous reports that the vvIBDV cluster can be defined based on both genome segments (Islam et al., 2001), which suggests a worldwide spread of a single vvIBDV clone and/ or an essential contribution of both genome segments to virus replication and virulence.
Although most significant clusters were maintained in the A- and B-derived phylogenies, 13 strains (26 %) were identified as exhibiting a significantly different phylogenetic position depending on which segment was analysed. Most strikingly, some strains exhibited markedly different genetic relatives for segments A and B. Sequencing the full coding sequence of both genome segments of strain 02015.1 confirmed that its segment A was phylogenetically related to the vvIBDV cluster, whereas its segment B was not. The demonstration of such a strain could suggest the existence of previously unrecognized segment B lineages that can be associated with a vvIBDV-like segment A and/or a possible reassortment between a vvIBDV (segment A) and a yet-tobe- identified parent with a different genetic background. In IBDV, several reports have proposed that natural reassortment is possible (Brown & Skinner, 1996; Yamaguchi et al., 1997; Sun et al., 2003; Kong et al., 2004); however, reassortment has not been assessed statistically on such a wide panel of strains as used in the present study. Chimeric IBDVs have been constructed in the laboratory, thus demonstrating the feasibility of reassortment of virus RNAs (Boot et al., 2000; Brandt et al., 2001; Liu & Vakharia, 2004; Zierenberg et al., 2004), but the nucleotide signals that control the compatibility of genome segments from different IBDV lineages (and thus acceptance of these segments by reassortant viruses) have not yet been identified. These mechanisms have been recently characterized in other dsRNA viruses with a segmented genome, the mammalian reoviruses. In these viruses, the incorporation of an engineered ssRNA into the genome depends critically on three consecutive nucleotides of the 59 UTR, and mutations at these positions most likely modify the secondary structure of the 59 UTR (Roner et al., 2004). The reverse-genetics system developed for IBDV (Mundt & Vakharia, 1996) provides a unique tool to perform a similar investigation in IBDV, and the UTRs of the genome segments of strain 02015.1 are currently under study.
The pathogenicity of the possibly reassortant strain 02015.1 was shown experimentally to be significantly lower than that of a typical vvIBDV, a result which might suggest that both segments might be involved in the pathogenicity of IBDV. The published literature regarding the influence of segment B on IBDV phenotype produces somewhat conflicting results: one study suggests that segment B has a limited influence on pathogenicity (Boot et al., 2000), whereas others have suggested that segment B could influence replication efficiency and modulate bursal lesions (Liu & Vakharia, 2004; Zierenberg et al., 2004; Boot et al., 2005). In the present study, the three pathogenic viruses induced the same virus titres and amounts of antigen in the bursae of challenged chickens. Hence, segments A and B of strain 02015.1 seem to be compatible, and the reduced mortality caused by 02015.1 does not seem to be due to inefficient replication of this virus. However, segment A of 02015.1 still has specific mutations (two amino acid mutations were identified in the coding region and the UTRs have not yet been sequenced). We therefore cannot be sure that segment A of the reassortant has retained the potential for hypervirulence. To answer this question, the recovery in vitro of a pathogenic virus generated by reverse genetics from segment A of 02015.1 combined with segment B of one or more vvIBDVs would be required to demonstrate that the atypical segment B of strain 02015.1 was indeed responsible for its reduced pathogenicity.
To conclude, the present study provides interesting insights into the evolution of vvIBDV. Indeed, there are increasing reports of vvIBDV-related strains that differ genetically and/or phenotypically from the vvIBDV reference strains isolated in the late 1980s. van den Berg et al. (2004) described the Henan strain, which exhibited a vVP2 phylogenetically related to the vvIBDV cluster but had reduced pathogenicity compared with typical vvIBDVs. The present study suggests that the Henan strain resembles 02015.1 with respect to reduced pathogenicity and possible reassortment. Others reports describe vvIBDV isolates from Indonesia and Malaysia with amino acid substitutions in the vVP2 region and which did not cause clinical disease or mortality (Hoque et al., 2001; Parede et al., 2003), whereas another report described a pathogenic vvIBDV strain with extensive antigenic changes due to amino acid substitutions in the vVP2 region (Eterradossi et al., 2004). Taken together, these observations suggest that genetic evolution from the initial vvIBDV clone has led to the emergence of new and diversified vvIBDV-related virus strains. The characterization of such emerging viruses calls for the definition of validated pathogenicity markers and for the development of diagnostic assays targeted at both genome segments.
REFERENCES
Bayliss, C. D., Spies, U., Shaw, K., Peters, R. W., Papageorgiou, A., Mu¨ ller, H. & Boursnell, M. E. (1990). A comparison of the sequences of segment A of four infectious bursal disease virus strains and identification of a variable region in VP2. J Gen Virol 71, 1303–1312.
Boot, H. J., ter Huurne, A. A., Hoekman, A. J., Peeters, B. P. & Gielkens, A. L. (2000). Rescue of very virulent mosaic IBDV from cloned cDNA: VP2 is not the sole determinant of the very virulent phenotype. J Virol 74, 6701–6711.
Boot, H. J., Hoekman, A. J. & Gielkens, A. L. (2005). The enhanced virulence of very virulent infectious bursal disease virus is partly determined by its B segment. Arch Virol 150, 137–144.
Brandt, M., Yao, K., Meihong, L., Heckert, R. A. & Vakharia, V. N. (2001). Molecular determinants of virulence, cell tropism, and pathogenic phenotype of infectious bursal disease virus. J Virol 75, 11974–11982.
Brown, M. D. & Skinner, M. A. (1996). Coding sequences of both genome segments of a European ‘very virulent’ infectious bursal disease virus. Virus Res 40, 1–15.
Bygrave, A. C. & Faragher, J. T. (1970). Mortality associated with Gumboro disease. Vet Rec 86, 758–759.
Chettle, N. J., Stuart, J. C. & Wyeth, P. J. (1989). Outbreaks of virulent infectious bursal disease in East Anglia. Vet Rec 125, 271–272.
Delmas, B., Kibenge, F. S. B., Leong, J. C., Mundt, E., Vakharia, V. N. & Wu, J. L. (2004). Birnaviridae. In Virus Taxonomy. Eighth Report of the International Committee on Taxonomy of Viruses, pp. 561–569. Edited by C. M. Fauquet, M. A. Mayo, J. Maniloff, U. Desselberger & L. A. Ball. London: Academic Press.
Domanska, K., Mato, T., Rivallan, G. & 7 other authors (2004). Antigenic and genetic diversity of early European isolates of Infectious bursal disease virus prior to the emergence of the very virulent viruses: early European epidemiology of Infectious bursal disease virus revisited? Arch Virol 149, 465–480.
Eterradossi, N., Picault, J. P., Drouin, P., Guittet, M., L’Hospitalier, R. & Bennejean, G. (1992). Pathogenicity and preliminary antigenic characterization of six infectious bursal disease virus strains isolated in France from acute outbreaks. Zentralbl Veterinarmed B 39, 683–691.
Eterradossi, N., Toquin, D., Rivallan, G. & Guittet, M. (1997a). Modified activity of a VP2-located neutralizing epitope on various vaccine, pathogenic and hypervirulent strains of infectious bursal disease virus. Arch Virol 142, 255–270.
Eterradossi, N., Rivallan, G., Toquin, D. & Guittet, M. (1997b). Limited antigenic variation among recent infectious bursal disease virus isolates from France. Arch Virol 142, 2079–2087.
Eterradossi, N., Arnauld, C., Toquin, D. & Rivallan, G. (1998). Critical amino acid changes in VP2 variable domain are associated with typical and atypical antigenicity in very virulent infectious bursal disease viruses. Arch Virol 143, 1627–1636.
Eterradossi, N., Arnaud, C., Tekaia, F. & 7 other authors (1999). Antigenic and genetic relationships between European very virulent infectious bursal disease viruses and an early West African isolate. Avian Pathol 28, 36–46.
Eterradossi, N., Gauthier, C., Reda, I. & 9 other authors (2004). Extensive antigenic changes in an atypical isolate of very virulent infectious bursal disease virus and experimental clinical control of this virus with an antigenically classical live vaccine. Avian Pathol 33, 423–431.
Farris, J. S., Ka¨ llersjo¨ , M., Kluge, A. G. & Bult, C. (1994). Testing significance of incongruence. Cladistics 10, 315–319.
Felsenstein, J. (1993). PHYLIP (phylogeny inference package), version 3.5c. Distributed by the author. Department of Genome Sciences, University of Washington, Seattle, USA.
Guindon, S. & Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52, 696–704.
Hoque, M. M., Omar, A. R., Chong, L. K., Hair-Bejo, M. & Aini, I. (2001). Pathogenicity of SspI-positive infectious bursal disease virus and molecular characterization of the VP2 hypervariable region. Avian Pathol 30, 369–380.
Ignjatovic, J. & Sapats, S. (2002). Confirmation of the existence of two distinct genetic groups of infectious bursal disease virus in Australia. Aust Vet J 80, 689–694.
Islam, M. R., Zierenberg, K. & Muller, H. (2001). The genome segment B encoding the RNA-dependent RNA polymerase protein VP1 of very virulent infectious bursal disease virus (IBDV) is phylogenetically distinct from that of all other IBDV strains. Arch Virol 146, 2481–2492.
Kibenge, F. S. B., Dhillon, A. S. & Russell, R. G. (1988). Biochemistry and immunology of infectious bursal disease virus. J Gen Virol 69, 1757–1775.
Kong, L. L., Omar, A. R., Hair-Bejo, M., Aini, I. & Seow, H. F. (2004). Sequence analysis of both genome segments of two very virulent infectious bursal disease virus field isolates with distinct pathogenicity. Arch Virol 149, 425–434.
Lasher, H. N. & Shane, S. M. (1994). Infectious bursal disease. World Poultry Sci J 50, 133–166.
Lecointre, G. L., Rachdi, P., Darlu, P. & Denamur, E. (1998). Escherichia coli molecular phylogeny using the incongruence length difference test. Mol Biol Evol 15, 1685–1695.
Le Noue¨ n, C., Rivallan, G., Toquin, D. & Eterradossi, N. (2005). Significance of the genetic relationships deduced from partial nucleotide sequencing of infectious bursal disease virus genome segments A or B. Arch Virol 150, 313–325.
Lim, B. L., Cao, Y., Yu, T. & Mo, C. W. (1999). Adaptation of very virulent infectious bursal disease virus to chicken embryonic fibroblasts by site-directed mutagenesis of residues 279 and 284 of viral coat protein VP2. J Virol 73, 2854–2862.
Liu, M. & Vakharia, V. N. (2004). VP1 protein of infectious bursal disease virus modulates the virulence in vivo. Virology 330, 62–73.
Lombardo, E., Maraver, A., Espinosa, I., Fernandez-Arias, A. &
Rodriguez, F. J. (2000). VP5, the nonstructural polypeptide of infectious bursal disease virus, accumulates within the host plasma membrane and induces cell lysis. Virology 277, 345–357.
Macreadie, I. & Azad, A. A. (1993). Expression and RNA dependent RNA polymerase activity of birnavirus VP1 protein bacteria and yeast. Biochem Mol Biol Int 6, 1169–1178.
Mu¨ ller, H. & Nitschke, R. (1987). The two segments of the infectious bursal disease virus genome are circularized by a 90,000-Da protein. Virology 159, 174–177.
Mundt, E. (1999). Tissue culture infectivity of different strains of infectious bursal disease virus is determined by distinct amino acids in VP2. J Gen Virol 80, 2067–2076.
Mundt, E. & Vakharia, V. N. (1996). Synthetic transcripts of doublestranded Birnavirus genome are infectious. Proc Natl Acad Sci U S A 93, 11131–11136.
Parede, L. H., Sapats, S., Gould, G., Rudd, M., Lowther, S. & Ignjatovic, J. (2003). Characterization of infectious bursal disease virus isolates from Indonesia indicates the existence of very virulent strains with unique genetic changes. Avian Pathol 32, 511–518.
Reed, L. J. & Muench, H. (1938). A simple method of estimation of fifty per cent end-points. Am J Hyg 27, 493–497.
Roner, M. R., Bassett, K. & Roehr, J. (2004). Identification of the 59 sequences required for incorporation of an engineered ssRNA into the Reovirus genome. Virology 329, 348–360.
Sapats, S. I., Ashton, F., Wright, P. J. & Ignjatovic, J. (1996). Sequence analysis of the S1 glycoprotein of infectious bronchitis viruses: identification of a novel genotypic group in Australia. J Gen Virol 77, 413–418.
Schnitzler, D., Bernstein, F., Mu¨ ller, H. & Becht, H. (1993). The genetic basis for the antigenicity of the VP2 protein of the infectious bursal disease virus. J Gen Virol 74, 1563–1571.
Skeeles, J. K., Luckert, P. D., Fletcher, O. J. & Leonard, D. J. (1979). Immunization studies with a cell culture adapted infectious bursal disease virus. Avian Dis 23, 456–465.
Spies, U., Mu¨ ller, H. & Becht, H. (1989). Nucleotide sequence of infectious bursal disease virus genome segment A delineates two major open reading frames. Nucleic Acids Res 17, 7982.
Sun, J. H., Lu, P., Yan, Y. X., Hua, X. G., Jiang, J. & Zhao, Y. (2003). Sequence and analysis of genomic segment A and B of very virulent infectious bursal disease virus isolated from China. J Vet Med B Infect Dis Vet Public Health 50, 148–154.
Swofford, D. L. (2002). PAUP* Phylogenetic Analysis Using Parsimony. Sunderland, MA: Sinauer Associates.
Tacken, M. G., Rottier, P. J., Gielkens, A. L. & Peeters, B. P. (2000). Interactions in vivo between the proteins of infectious bursal disease virus: capsid protein VP3 interacts with the RNA-dependent RNA polymerase, VP1. J Gen Virol 81, 209–218.
Tacken, M. G., Peeters, B. P., Thomas, A. A., Rottier, P. J. & Boot, H. J. (2002). Infectious bursal disease virus capsid protein VP3 interacts both with VP1, the RNA-dependent RNA polymerase, and with viral double-stranded RNA. J Virol 76, 11301–11311.
Vakharia, V. N., He, J., Ahamed, B. & Snyder, D. B. (1994). Molecular basis of antigenic variation in infectious bursal disease virus. Virus Res 31, 265–273.
van den Berg, T. P., Gonze, M. & Meulemans, G. (1991). Acute infectious bursal disease in poultry: isolation and characterisation of a highly virulent strain. Avian Pathol 20, 409–421.
van den Berg, T. P., Morales, D. N., Eterradossi, N. & 12 other authors (2004). Assessment of genetic, antigenic and pathotypic criteria for the characterization of IBDV strains. Avian Pathol 33, 470–476.
van Loon, A. A. W. M., de Haas, N., Zeyda, I. & Mundt, E. (2002). Alteration of amino acids in VP2 of very virulent infectious bursal disease virus results in tissue culture adaptation and attenuation in chickens. J Gen Virol 83, 121–129.
von Einem, U. I., Gorbalenya, A. E., Schirrmeier, H., Behrens, S. E., Letzel, T. & Mundt, E. (2004). VP1 of infectious bursal disease virus is an RNA-dependent RNA polymerase. J Gen Virol 85, 2221–2229.
Yamaguchi, T., Ogawa, M., Miyoshi, M., Inoshima, Y., Fukushi, H. & Hirai, K. (1997). Sequence and phylogenetic analyses of highly virulent infectious bursal disease virus. Arch Virol 142, 1441–1458.
Yao, K. & Vakharia, N. V. (2001). Induction of apoptosis in vitro by the 17-kDa nonstructural protein of infectious bursal disease virus: possible role in viral pathogenesis. Virology 285, 50–58.
Yao, K., Goodwin, M. A. & Vakharia, V. N. (1998). Generation of a mutant infectious bursal disease virus that do not cause bursal lesions. J Virol 72, 2647–2654.
Zierenberg, K., Nieper, H., van den Berg, T. P., Ezeokoli, C. D., Voss, M. & Muller, H. (2000). The VP2 variable region of African and German isolates of infectious bursal disease virus: comparison with very virulent, ‘‘classical’’ virulent, and attenuated tissue cultureadapted strains. Arch Virol 145, 113–125.
Zierenberg, K., Raue, R., Nieper, H., Islam, M. R., Eterradossi, N., Toquin, D. & Mu¨ ller, H. (2004). Generation of serotype 1/serotype 2 reassortant viruses of the infectious bursal disease virus and their investigation in vitro and in vivo. Virus Res 105, 23–34.

